視頻報道
-

椎管內(nèi)囊腫病例:藏在脊柱中的惡魔
CCTV-4《中華醫(yī)藥》
-

突出的腫瘤-腦膜瘤
CCTV10《走進科學》
相關(guān)文章
Rasmussen’s encephalitis: clinical features and mechanisms advances
2016-12-13 12:05 作者:三博腦科醫(yī)院
Tianfu Li,Qing Gao, Guoming Luan
Epilepsy center, Sanbo Brain Hospital, Capital Medical University, Beijing, China.
Abstract:
Rasmussen’s encephalitis (RE) is neurological disorder of childhood characterized by uni-hemispheric inflammation, intractable focal epilepsy and progressive cognitive and neurological deficits. Currently, hemispherectomy is the only effective method to date to control the seizures associated with RE. Although this disease has been heavily investigated, the pathogenesis of RE with unilateral cortex atrophy and focal seizure is still enigmatic. Neuropathological and immunological studies support the hypothesis that destruction of neurons and astrocytes by cytotoxic CD8 T cells as a pathogenic mechanism underlying this enigmatic disorder. Recently data indicated that intrinsic activation of endogenous pro-inflammation high-mobility group box-1 (HMGB1) and Toll-like receptor (TLR) pathways, and dysregulation of adenosinergic mechanism are involved in the development of epilepsy, which suggest the specific targets in the treatment of epilepsy, inflammation and cognitive deterioration associated with epilepsy in RE patients.
Key words: Rasmussen encephalitis, epilepsy, inflammation, HMGB1, adenosine, adenosine kinase
Introduction
Rasmussen’s encephalitis (RE) was initially described by neurosurgeon Theodore Rasmussen and his colleagues from the Montreal Neurological Institute in the late 1950s (RASMUSSEN et al., 1958). From then on, extensive research on the clinical features, neuropathology,mechanisms and therapy of RE was carried out, and the 2005 European consensus on pathogenesis, diagnosis,and treatment of RE was achieved and still remains accepted guideline for evaluative criteria (Bien et al., 2005;Olson et al., 2013). RE is a very rare chronic progressive inflammatory neurological disorder of uncertain etiology affecting mostly children and associated with hemispheric atrophy, pharmacoresistant focal epilepsy (epilepsia partialis continua), cognitive deterioration and progressive cognitive and neurological deficits, resulting from progressive loss of function subserved by the involved cerebral hemisphere (Bien et al., 2005; Bien et al., 2002; RASMUSSEN et al.,1958; Varadkar et al., 2014). An intriguing feature of RE is the restriction of the inflammatory process to one brain hemisphere, setting it apart from any other inflammatory disease of the CNS. The aetiology and pathogenesis of RE,in particular, the factors responsible for the characteristic of asymmetry are still elusive. The neuropathological hallmarks of RE consist of lymphocytic infiltrates (perivascular lymphocytic cuffing), microglial nodules, neuronal destruction, and gliosis of the affected hemisphere (Farrell et al., 1995; Rogers et al., 1994). Currently, targeted therapeutic strategies remain elusive and hemispherectomy is the only effective method to control the seizures associated with RE.Recent results demonstrate that activation of endogenous high-mobility group box-1 (HMGB1) and Toll-like receptor (Luan et al., 2016), and adenosine system dysfunction in epileptic tissue (Luan et al., 2013), which may play a role in the generation of seizures, and possibly epileptogenesis itself in RE. Here, we will review the current basic and clinical research associated with RE patients.
Pathogenesis of RE
Over fifty years, extensive studies have been carried out to attempt to elucidate the pathogenesis of RE. The main hypothesis on the pathogenesis of RE as follows: i) virus infection. The hypothesis on virus infection in RE suggested by Rasmussen, is that RE may be associated with constituents of the immune reaction in the brain following a virus infection, such as lymphocyte infiltration and microglial nodules. Support of Rasmussen’s idea comes from studies linking cytomegalovirus and herpes simplex virus (RASMUSSEN et al., 1958). Nevertheless, using the method of polymerase chain reaction (PCR) and/ or in situ hybridization, results from other research groups demonstrate that the virus DNA sequence existed within the tissue of epilepsy patients associated with other central nervous system (CNS) diseases such as focal cortical dysplasia, gangliocytoma and cerebromalacia (Jay et al., 1995; Rogers et al., 1994). Several viruses, including Epstein-Barr virus, cytomegalovirus, herpes simplex virus, and enterovirus has been tested for the presence in brain tissue from patients with RE (Friedman et al., 1977; Power et al., 1990; Walter and Renella, 1989). None of these studies was able to show a causal association between Rasmussen’s encephalitis and a specific virus. To establish a reliable method to identify the specific virus in RE is challenging, for infected cells under cytotoxic T-cell attack can be injured and later engulfed by microglia and macrophages, in which case the chances of finding a virus most likely occur early in the disease when surgical sampling is less frequent (Varadkar et al., 2014). ii) Antibody-mediated CNS degeneration. Anti-glutamate receptors GluR3, was the first circulating auto-antibodies identified in RE in 1994 (Rogers et al., 1994). From then on, antibody against GluR3, munc-18 and NMDA receptor-mediated mechanisms dominated RE research for more then 10 years (Alvarez- Baron et al., 2008; Granata et al., 2003; Greiner et al., 2011; Mantegazza et al., 2002; Rogers et al., 1994; Watson et al., 2005; Watson et al., 2004; Wiendl et al., 2001; Yang et al., 2000). However, the follow-up studies demonstrate that the circulating auto-antibodies, even those highly-specific cell surface-directed antibodies can occasionally be present in patients with neurodegenerative disease, in which case these antibodies are probably secondary to the pathology rather than causative. None of the autoantibodies have been found in more than a small number of patients with RE, and responses to plasma exchange are unpredictable. Therefore, the role of circulating autoantibodies in the pathogenesis of RE is still elusive (Varadkar et al., 2014). iii) T-cell cytotoxicity. Recent studies have suggested that in contrast to a random attraction of cells as part of a secondary immune response, CD8+ T cell-mediated attack against neurons and astrocytes in the central nervous system is a major component of the pathogenesis of RE (Bauer et al., 2007;Bien et al., 2002; Schwab et al., 2009). Because neurons and astrocytes are attacked by cytotoxic T cells, there may be an autoantigen, as a driving antigen expressed by both neurons and astrocytes. However, to identity the driving antigen of cytotoxic T cells is still elusive (Varadkar et al.,2014).
Advance of Pathogenesis
Dysfunction of adenosine system in RE
Adenosine is an endogenous purine nucleoside that modulates a wide range of physiological functions (Fredholm et al., 2011). Most notable among its many roles is its importance in controlling inflammation (Fredholm et al., 2011;Blackburn et al., 2009; Mills et al., 2012; Wen et al., 2010) and inhibiting seizures (Li et al., 2012; Li et al., 2009; Li et al.,2008; Li et al., 2007; Masino et al., 2011; Wilz et al., 2008) and restore cognitive function related with epilepy (Boison,2016). Recent evidence illustrated that the adenosine dysfunction in astrogliosis including decrease in the density of A1Rs (Glass et al., 1996). Extracellular levels of adenosine are regulated largely by an astrocyte-based adenosine cycle and astrocytic ADK is the major adenosine-removing enzyme (Boison et al., 2010). Minor changes in ADK activity translate rapidly into major changes in adenosine (Boison,2006). Therefore, upregulation of ADK in astrogliosis leads to reducing the“tone"of ambient adenosine leading to insufficient activation of adenosine receptors (Boison, 2008).Neuronal excitability in the brain is modulated by activation of G protein coupled adenosine receptors (A1, A2A, A2B, A3) (Fredholm et al., 2005; Fredholm et al., 2011). The receptor expression levels and availability of endogenous adenosine to activate the receptors plays a crucial role in neuronal excitability (Boison, 2016). Adenosine is a neuromodulator that has been proved to be major endogenous anticonvulsant acting via A1R. In the brain, adenosine modulates neuronal activity by decreasing presynaptic release of various neurotransmitters, and the most dramatic inhibitory actions are on the glutamatergic system (Reppert et al., 1991).In addition, adenosine acting through postsynaptic A1Rs may activate K+ channels, leading to hyperpolarization of postsynaptic neurons and promoting NMDA receptor inhibition (Wardas, 2002).
Recent work demonstrated that increased expression of major adenosine-removing enzyme adenosine kinase (ADK) in RE patients plays an important role in the epileptogensis of RE (Luan et al., 2013). Focal astrogliosis and marked expression of ADK were observed in the lesions of RE. Significantly greater ADK expression in RE versus controls was demonstrated by Western blot, and greater enzymatic activity for ADK was demonstrated using an enzymecoupled bioluminescent assay. ADK has been highlighted as a diagnostic marker to predict epileptogenesis as well as a potential target for anti-epileptogenesis or disease modification (Masino et al., 2011; Aronica et al., 2013;Boison, 2012; Boison, 2013; Boison, 2016; Li et al., 2012;Li et al., 2007; Li et al., 2008; Luan et al., 2015). Therefore,upregulation of ADK in RE is considered as a common pathologic hallmark of RE and that ADK might be a target in the treatment of epilepsy associated with RE (Luan et al., 2013). Neuronal adenosine A1 receptor (A1R) in the lesion area of RE was also observed (unpublished data).Activation of A1R has been proved to prevent the spatial spread of seizures (Fedele et al., 2006; Glass et al., 1996;Hamil et al., 2012; Klaft et al., 2016; Kochanek et al., 2006;Li et al., 2008; Masino et al., 2011; Wagner et al., 2010).Activation of A1R signaling maybe related to the clinical features of the RE: i) unilateral epileptic discharge in RE patients with or without unihemispheric slowing in long-term video EEG, and EPC is not always accompanied by visually recognisable ictal surface EEG activity; ii) both seizures and inflammation atrophy are seen only in one cerebral hemisphere, rarely spreading to the contrahemisphere,only four out of the roughly 200–300 published cases of Rasmussen’s encephalitis had evidence of bilateral disease on histopathology; iii) most of the clinical seizure types are focal seizures (SPS or CPS) with or without EPC, and notably, secondary generalized epilepsia continua from EPC has rarely been reported. Thereby, adenosine augmenting therapeutic strategies with anticonvulsion, anti-inflammation, and anti-cognitive dysfunction effects might be the ideal treatment for RE.
Activation of HMGB1-TLR-RAGE signaling in RE
Novel evidence of intrinsic activation of pro-inflammation signaling, endogenous high-mobility group box-1 (HMGB1),toll-like receptor (TLR) and RAGE in human Rasmussen’s encephalitis has been reported recently (Luan et al., 2016).HMGB-1) is a 29 kDa DNA-binding protein with a highly conserved structure in several species (Thomas, 2001).HMGB-1 participates in nucleosome formation and regulation of gene transcription (Stros et al., 2002; Park et al., 2003),including proinflammatory genes (Bianchi and Manfredi,2009). In response to inflammatory stimuli, HMGB-1 is secreted by activated macrophages (Bonaldi et al., 2003),natural killer cells (Semino et al., 2005), myeloid dendritic cells (Lotze and Tracey, 2005) and astrocytes (Maroso et al., 2010) binding to the receptor for advanced glycation end products (RAGE) and other receptors, including TLR2 and TLR4 (Scaffidi et al., 2002; Parker et al., 2004). HMGB1 acts as a “danger signal” and alerts the immune system to damaged or dying cells. The hyperacetylated form of HMGB1 regulates transcription of various pro-inflammatory cytokines through binding to TLR2, TLR4 and also to RAGE (Maroso et al., 2011; Bianchi and Manfredi, 2009). HMGB1-TLR-RAGE may represent a novel pro-inflammatory axis following sterile brain injury (Walker and Sills, 2012).
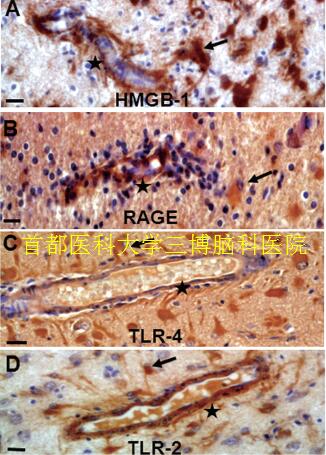
▲Fig.1. HMGB-1, RAGE, TLR-4 and TLR-2 immunoreactivity in RE.Nuclear and cytoplasmic staining showed in perivascular glial cells(A, arrows) and marked cytoplasmic staining of RAGE (B, arrows),TLR-4(C, arrows), TLR-2 (D, arrows). Endothelial cells within the lesions cortex displayed HMGB1 (A, stars), RAGE (B, stars), TLR-4 (C, stars) and TLR-2 immunoreactivity (D, stars). Scale bar: 12.5μm.
In addition to exert a pro-inflammatory role, HMGB1-TLRRAGE signaling plays a critical role in determining the pathological outcomes epilepsy (Iori et al., 2013; Maroso et al., 2010; Riazi et al., 2010; Rodgers et al., 2009; Zurolo et al., 2011). Although HMGB1 release and signaling may be a general feature of all epilepsies (Iori et al., 2013; Maroso et al., 2010; Zurolo et al., 2011), expression of HMGB1,TLR2, TLR4 and RAGE was more markedly increased in perivascular areas, and endothelial cells in walls of blood vessels within the lesions cortex displayed immunoreactivity in RE patients (Fig.1) (Luan et al., 2016). These findings are concomitant with the reactive astrogliosis, neuron loss and inflammation (i.e. CD8-positive, CD3-positive T lymphocyte).The evidence further supported the role of HMGB1-TLR pathway in activation of immune and endothelial cells in the pathogenesis of RE. Anti-inflammatory agents targeting on HMGB1-TLRs-RAGE may also prove beneficial in alleviating some of the common comorbidities associated with chronic epilepsy, including cognitive dysfunction and memory deficits (Costello et al., 2011; Mazarati et al., 2011; Vezzani et al.,2013), which will provide benefits for RE in three ways: antiinflammtion, aniti-epilepsy and improve cognitive dysfunction associated with epilepsy (Luan et al., 2016).
Clinical features
Rasmussen’s encephalitis is a progressive disease characterized by drug-resistant focal epilepsy, progressive hemiplegia, and cognitive deterioration, with unihemispheric brain atrophy (Bien et al., 2002; Hoffman et al., 2016; Olson et al., 2013; Pulsifer et al., 2004; Varadkar et al., 2014).The disorder is rare and affects mostly children or young adults, and the median age of onset is 6 years and the range from infancy to adulthood (Bien et al., 2002; Granata et al.,2003a). The typical clinical course of RE has been proposed last century (Bien et al., 2002). Stage 1 (pordromal phase) is characterized by a non-specific low seizure frequency and, in rare instances, some degree of hemiparesis with a average duration of 7.1 months (range 0 to 8.1 years) and even longer duration in adolescent and adult patients compared with the children. Stage 2 (acute stage) is characterized by frequent simple partial motor seizures, and EPC in 69% of cases. The median duration of this stage was 8 months (range 4-8 months). During the acute stage the neurological deficits appeared including progressive hemiparesis,cognitive deterioration, and aphasia if dominant hemisphere affected. The last stage (residual stage) is characterized by continuing seizures with a marked decrease in seizure frequency, notably, with permanent and stable neurological deficit (Bien et al., 2002).
EEG, neuroimaging of RE
Overall, no specific EEG changes identified to differentiate RE from other causes of focal epilepsy such as focal cortical dysplasia (Longaretti et al., 2012). Long-term video EEG results were abnormal in all cases and generally expressed slow waves in the affected hemisphere. Polymorphic delta waves and multifocal unilateral epileptic discharge interictal EEG were mainly observed in temporal and central locations.In the majority of patients, contralateral asynchronous slow waves and epileptiform discharges occurred (Granata et al.,2003b). However, few ictal patterns were ever recorded from contralateral electrodes (Andrews et al., 1997). Up to now,only four patents have been reported to have bilateral RE (Guan et al., 2011). Epileptiform abnormalities are frequent and they often develop into electrographic seizures, however, EPC in RE is not always accompanied by rhythmic EEG discharges on surface EEG (Varadkar et al., 2014). Emerging persistent delta activity over the affected hemisphere with contralateral normal background rhythms, followed in due course by independent interictal epileptiform abnormalities over the unaffected hemisphere highly indicate the diagnosis of RE as the condition evolves, and highlight as the marker of overall cognitive deterioration (Longaretti et al., 2012). Magnetic resonance imaging (MRI) of the brain has become a key tool for diagnostic assessment, especially provides the evidence of the diagnosis in the early stages and the progression of RE (Chiapparini et al., 2003). MRI findings of RE patients usually indicate a progressive unihemispheric focal cortical atrophy with mild or severe enlargement of the lateral ventricle (Fig.2). The majority of patients exhibit unilateral enlargement of the inner and outer CSF compartments in the insular and periinsular regions during the early stage (Chiapparini et al., 2003; Chinchilla et al.,1994). In majority of RE patients, ipsilateral moderate atrophy of the head of the caudate nucleus is a typical alteration of MRI and regarded as an sign for early stage of RE, but not an invariable accompanying feature of hemispheric atrophy(Chiapparini et al., 2003). Even in the early stage of RE, normal findings of MRI scans or scans with gadolinium enhancement are very rare in RE patients (Bien et al., 2002;Chiapparini et al., 2003; Kaiboriboon et al., 2000; Lee et al., 2001). Functional studies using18F-FDG positron emission tomography (PET) illustrates diffuse unilateral cerebral hypometabolism within the affected hemisphere (Fiorella et al., 2001).
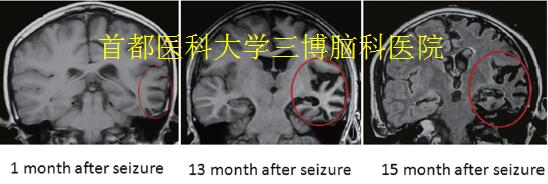
▲Fig.2. Neuroimaging in RE
(A-C): The brain MRI displayed a typical progressive abnormal signal area expansion and atrophy of the left hemisphere, with mild enlargement of the left lateral ventricle (1 months, 13 months, 15months after the first seizure onset).
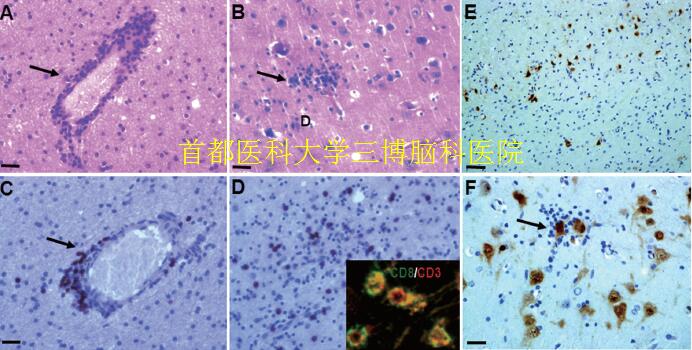
▲Fig.3. Neuropathological features in RE
(A): Perivascular lymphocytic cuffing in the cerebral cortex (arrows, HE staining). (B): formation of microglial nodules and diffuse microglial activation (arrows, HE staining). (C): Perivascular lymphocytic cuffing in the cerebral cortex (arrows, CD8 staining). (D): parenchymal lymphocytic in the cerebral cortex (arrows, CD8 staining). Inset in D: co-localization of CD3 and CD8. (E): neuronal loss in the lesion cortex of RE (NeuN staining). (F): Cortical neuron with neuronophagia (arrows, NeuN staining). Scale bar: A, B, C, D, F: 25μm; E:100μm.
Characteristic of neuropathology in RE
The neuropathologic hallmarks of RE include inflammation (perivascular lymphocytic cuffing, microglial nodules, leptomeningeal infiltrates), neuronal loss, and astrogliosis confined to one cerebral hemisphere (Fig.3) (Farrell et al., 1995; Pardo et al., 2004; RASMUSSEN et al., 1958; Rogers et al., 1994). Microglial and lymphocytic nodules and perivascular cuffing, neuronal death, and neuronophagia are the most common pathological features. T cells aggregating around the perivascular and infiltrating the meninges are as the majority of CD3+CD8+ cells. During the residual stage the pathological features include cortical cavitation, marked astrogliosis, and neuronal cell loss. Recently evidence illustrates dual pathology, with the finding of focal cortical dysplasia, tuberous sclerosis, low grade tumor, vascular abnormalities or old ischaemic lesions in association with Rasmussen’s encephalitis (Bien et al., 2002; Hart et al.,1998; Iyer et al., 2010; Palmer et al., 1999; Takei et al., 2010;Thom et al., 1999). Inflammation is usually multifocal within the hemisphere and progressive, with the characteristic of an area of pronounced cortical damage is often surrounded by normal cerebral cortex or milder stages of inflammation,which illustrate the reason why a biopsy results sometimes mislead in the diagnosis (Varadkar et al.,2014).
Acknowledgment
This Project was supported by the Grant from the BIBDPXM2013_014226_07_000084, National Natural Science Foundation of China (81571275), Scientific Research Common Program of Beijing Commission of Education(KM201410025027), Beijing Municipal Natural Science Foundation (7144217) and Beijing Municipal Science & Technology Commission (Z131107002213171). We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Competing interests
The authors declare that they have no competing interests.
 京公網(wǎng)安備 11010802035500號
京公網(wǎng)安備 11010802035500號